Abstract
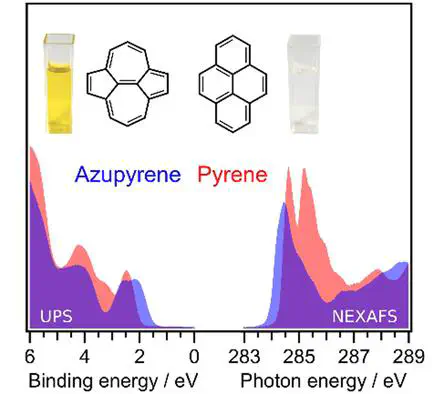
Pyrene derivatives play a prominent role in organic electronic devices, including field effect transistors, light emitting diodes, and solar cells. The flexibility in the desired properties has previously been achieved by variation of substituents at the periphery of the pyrene backbone. In contrast, the influence of the topology of the central π-electron system on the relevant properties such as the band gap or the fluorescence behavior has not yet been addressed. In this work, pyrene is compared with its structural isomer azupyrene, which has a π-electron system with non-alternant topology. Using photoelectron spectroscopy, near edge X-ray absorption fine structure spectroscopy, and other methods, it is shown that the electronic band gap of azupyrene is by 0.72 eV smaller than that of pyrene. The difference of the optical band gaps is even larger with 1.09 eV, as determined by ultraviolet–visible absorption spectroscopy. The non-alternant nature of azupyrene is also associated with a more localized charge distribution. Further insight is provided by density functional theory (DFT) calculations of the molecular properties and ab initio coupled cluster calculations of the optical transitions. The concept of aromaticity is used to interpret the major topology-related differences.
Samuel J. Hall
Postdoctoral Asisstant
Computational chemist specialising in electronic structure calculations and machine learning for core-level x-ray spectroscopy simulations.