The Nuts and Bolts of Core-Hole Constrained Ab-Initio Simulation for K-Shell X-ray Photoemission and Adsorption Spectra
Abstract
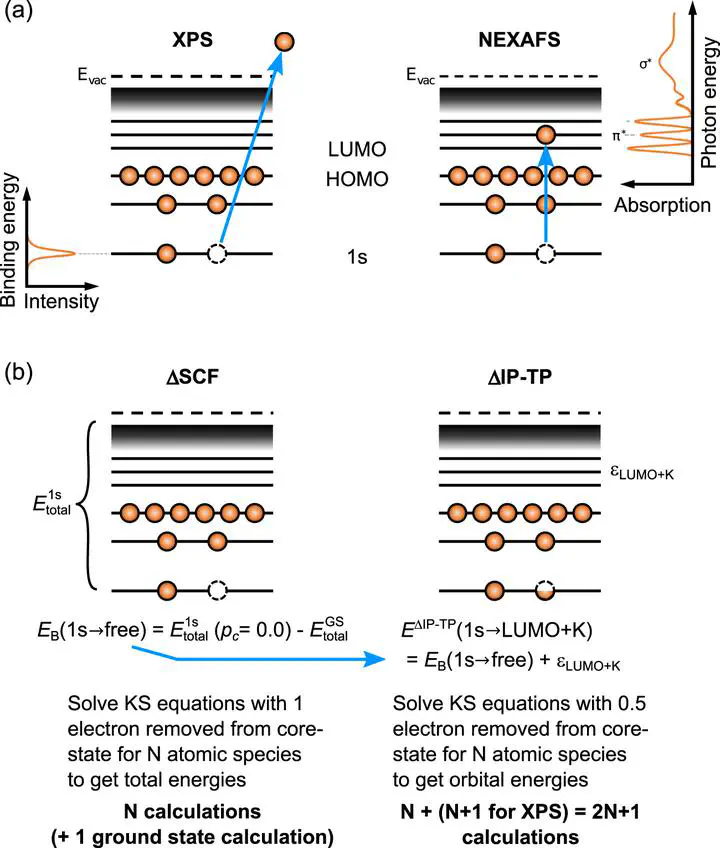
X-ray photoemission (XPS) and near edge x-ray absorption fine structure (NEXAFS) spectroscopy play an important role in investigating the structure and electronic structure of materials and surfaces. Ab initio simulations provide crucial support for the interpretation of complex spectra containing overlapping signatures. Approximate core-hole simulation methods based on density functional theory (DFT) such as the delta-self-consistent-field (ΔSCF) method or the transition potential (TP) method are widely used to predict K-shell XPS and NEXAFS signatures of organic molecules, inorganic materials and metal-organic interfaces at reliable accuracy and affordable computational cost. We present the numerical and technical details of our variants of the ΔSCF and TP method (coined ΔIP-TP) to simulate XPS and NEXAFS transitions. Using exemplary molecules in gas-phase, in bulk crystals, and at metal-organic interfaces, we systematically assess how practical simulation choices affect the stability and accuracy of simulations. These include the choice of exchange-correlation functional, basis set, the method of core-hole localization, and the use of periodic boundary conditions (PBC). We particularly focus on the choice of aperiodic or periodic description of systems and how spurious charge effects in periodic calculations affect the simulation outcomes. For the benefit of practitioners in the field, we discuss sensible default choices, limitations of the methods, and future prospects.
Samuel J. Hall
Postdoctoral Asisstant
Computational chemist specialising in electronic structure calculations and machine learning for core-level x-ray spectroscopy simulations.