The Molecule-Metal Bond of Alternant versus Nonalternant Aromatic Systems on Coinage Metal Surfaces: Naphthalene versus Azulene on Ag(111) and Cu(111)
Abstract
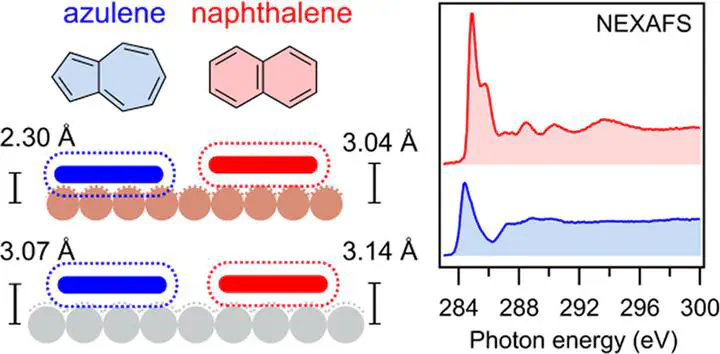
Interfaces between polycyclic π-electron systems and metals play prominent roles in organic or graphene-based (opto)electronic devices, in which performance-related parameters depend critically on the properties of metal/semiconductor contacts. Here, we explore how the topology of the π-electron system influences the bonding and the electronic properties of the interface. We use azulene as a model for nonalternant pentagon–heptagon (5–7) ring pairs and compare it to its isomer naphthalene, which represents the alternant 6–6 ring pair. Their coverage-dependent interaction with Ag(111) and Cu(111) surfaces was studied with the normal-incidence X-ray standing wave (NIXSW) technique, near-edge X-ray absorption fine structure (NEXAFS) spectroscopy, UV and X-ray photoelectron spectroscopies (UPS and XPS), and density functional theory (DFT). Coverage-dependent adsorption heights and spectroscopic data reveal that azulene forms shorter interfacial bonds than naphthalene and engages in stronger electronic interactions with both surfaces. These differences are more pronounced on Cu. Increasing coverages lead to larger adsorption heights, indicating bond weakening by intermolecular repulsion. The extensive DFT calculations include dispersive interactions using (1) the DFT-D3 scheme, (2) the vdWsurf correction based on DFT-TS, (3) a many-body dispersion (MBD) correction scheme, and (4) the D3surf scheme. All methods predict the adsorption heights reasonably well with an average error below 0.1 Å. The stronger bond of azulene is attributed to its nonalternant topology, which results in a reduced highest occupied molecular orbital (HOMO)–lowest occupied molecular orbital (LUMO) gap and brings the LUMO energetically close to the Fermi energy of the metal, causing stronger hybridization with electronic states of the metal surfaces.
Samuel J. Hall
Postdoctoral Asisstant
Computational chemist specialising in electronic structure calculations and machine learning for core-level x-ray spectroscopy simulations.