Enhanced Bonding of Pentagon–Heptagon Defects in Graphene to Metal Surfaces: Insights From the Adsorption of Azulene and Naphthalene to Pt(111)
Abstract
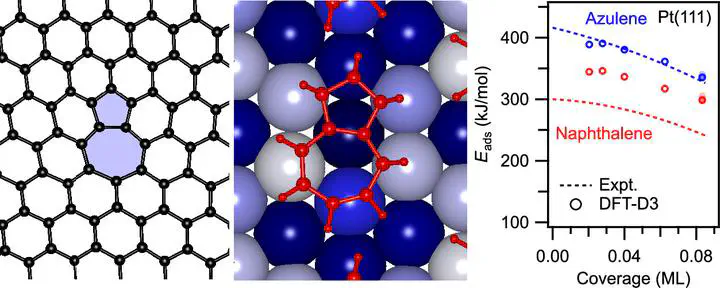
The performance of graphene-based (opto)electronic devices depends critically on the graphene/metal interface formed at the metal contacts. We show here that the interface properties may be controlled by topological defects, such as the pentagon–heptagon (5–7) pairs, because of their strongly enhanced bonding to the metal. To measure the bond energy and other key properties not accessible for the embedded defects, we use azulene as a molecular model for the 5–7 defect. Comparison to its isomer naphthalene, which represents the regular graphene structure, reveals that azulene interacts more strongly with a Pt(111) surface. Its adsorption energy, as measured by single-crystal adsorption calorimetry (SCAC), exceeds that of naphthalene by up to 116 kJ/mol (or up to 50%). Both isomers undergo hybridization of their frontier orbitals with metal states, as indicated by X-ray and ultraviolet photoelectron spectroscopy (XPS/UPS) and near-edge X-ray absorption fine structure (NEXAFS) spectroscopy combined with molecular orbital (MO) projection analysis through dispersion-corrected, periodic density functional theory (DFT) calculations. Based on the NEXAFS/DFT analysis, the stronger bond of the 5–7 system is attributed to the different energetic response of its unoccupied frontier orbitals to adsorption. Adsorption-induced bond-length changes show substantial topology-related differences between the isomers. Electron transfer occurs in both directions through donation/back-donation, resulting in the partial occupation (deoccupation) of formerly unoccupied (occupied) orbitals, as revealed by periodic energy decomposition analysis (pEDA) for extended systems. Our model study shows that the topology of the π-electron system strongly affects its bonding to a transition metal and thus can be utilized to tailor interface properties.
Samuel J. Hall
Postdoctoral Asisstant
Computational chemist specialising in electronic structure calculations and machine learning for core-level x-ray spectroscopy simulations.