Characterizing Molecule–Metal Surface Chemistry with Ab Initio Simulation of X-ray Absorption and Photoemission Spectra
Abstract
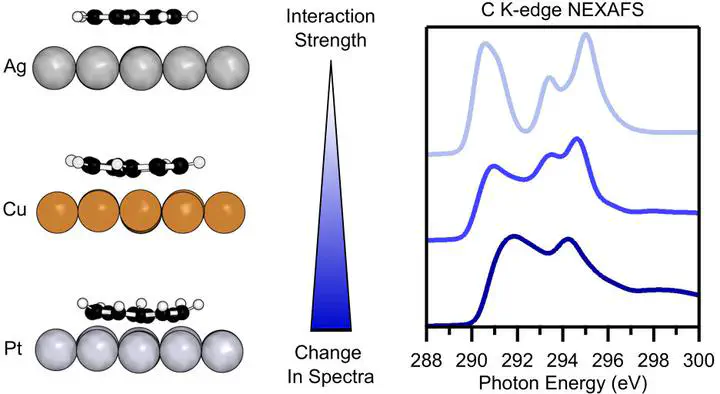
X-ray photoemission and X-ray absorption spectroscopy are important techniques to characterize chemical bonding at surfaces and are often used to identify the strength and nature of adsorbate–substrate interactions. In this study, we judge the ability of X-ray spectroscopic techniques to identify different regimes of chemical bonding at metal–organic interfaces. To achieve this, we sample different interaction strength regimes in a comprehensive and systematic way by comparing two topological isomers, azulene and naphthalene, adsorbed on three metal substrates with varying reactivity, namely the (111) facets of Ag, Cu, and Pt. Using density functional theory, we simulate core-level binding energies and X-ray absorption spectra of the molecular carbon species. The simulated spectra reveal three distinct characteristics based on the molecule-specific spectral features which we attribute to types of surface chemical bonding with varying strength. We find that weak physisorption only leads to minor changes compared to the gas-phase spectra, weak chemisorption leads to charge transfer and significant spectral changes, and strong chemisorption leads to a loss of the molecule-specific features in the spectra. The classification we provide is aimed at assisting interpretation of experimental X-ray spectra for complex metal–organic interfaces.
Samuel J. Hall
Postdoctoral Asisstant
Computational chemist specialising in electronic structure calculations and machine learning for core-level x-ray spectroscopy simulations.